Alport syndrome is an inherited disease characterized by progressive loss of kidney function, hearing loss, and eye abnormalities. The syndrome is caused by genetic mutations that affect expression and/or function of the type IV collagen family protein isoforms COL4A3, COL4A4, and COL4A5, which help form the glomerular basement membranes.
 The onset, symptoms, progression, and severity of the disease vary greatly among Alport patients. One individual may exhibit a mild, slowly progressive form of the disorder, while even a sibling may have an earlier onset with severe complications.
The onset, symptoms, progression, and severity of the disease vary greatly among Alport patients. One individual may exhibit a mild, slowly progressive form of the disorder, while even a sibling may have an earlier onset with severe complications.
A new study by researchers in the Division of Nephrology at Washington University School of Medicine and the Department of Pathology and Laboratory Medicine at Rutgers University gives us new insight into one potential cause of the variation in the Alport syndrome phenotype.

Jeff Miner, PhD, FASN
The research group, headed by Jeffrey Miner, PhD, investigated a specific missense mutation in laminin β2 (LAMB2) found in a young patient with a mild version of Pierson syndrome, another genetic disease that affects eye and kidney function. The laminin protein, like collagen IV, is an integral component of the glomerular basement membrane. The mutation (LAMB2-S80R) was presumed to be pathogenic.
However, when the analogous murine mutation (Lamb2-S83R) was bioengineered into mice, the mice did not develop kidney disease. This led the researches to theorize that the LAMB2-S80R allele exacerbates kidney stress, or that the young patient had an additional mutation.
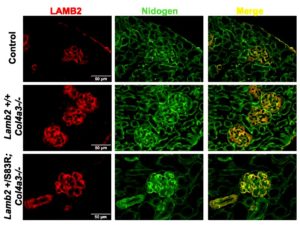
When the researchers bred just one copy of the analogous murine mutation (LAMB2-S83R) onto the Alport mouse background (absence of COL4A3) to stress the kidneys, the mice had a more rapidly progressive kidney disease and shorter longevity than typical Alport mice. In a milder model of X-linked Alport syndrome, in which female mice with one mutant copy of Col4A5 exhibit mosaic COL4A5 expression, the addition of the Lamb2-S83R resulted in increased proteinuria. Thus, mutations in non-collagen genes may worsen the severity of Alport syndrome.

Steven Funk
“Though initially unfruitful, our persistent investigation of the human LAMB2-S80R mutation gave us a 3-for-1 payout,” says Postdoc Research Associate and first author, Steven Funk. “Crossing the murine Lamb2-S83R allele with a mouse model of Alport syndrome revealed a potential mechanism of pathogenicity underlying the patient’s disease and additionally provided new insight into the highly variable spectrum of the Alport syndrome phenotype. Lastly, the CRISPR-mediated gene editing strategy produced a second Lamb2 mutation that represents a novel model of nephrotic syndrome unique in both its longevity and timing of foot process effacement.”
The study, Pathogenicity of a Human Laminin β2 Mutation Revealed in Models of Alport Syndrome is published online in the Journal of the American Society of Nephrology. Authors: Steven Funk, Raymond Bayer, Andrew Malone, Karen K. McKee, Peter D. Yurchenco, and Jeffrey Miner.